


How to CATCH your gene of interest
If you haven’t heard about CATCH by now, it’s time to catch up. Short for Cas9-assisted targeting of chromosome segments, CATCH comes from the lab of Yuval Ebenstein at Tel Aviv University and was first reported in this Nature Communications paper.
Like so many scientists, Ebenstein found himself routinely having to sequence whole-genome data in order to study a region that was too large to amplify easily with PCR. “You end up paying for all this data and eventually using a very small fraction of it,” he recalls. While there are several target-capture and enrichment methods, they all require knowledge of the sequence of interest. But for Ebenstein, who was interested in highly repetitive DNA, those methods didn’t work.
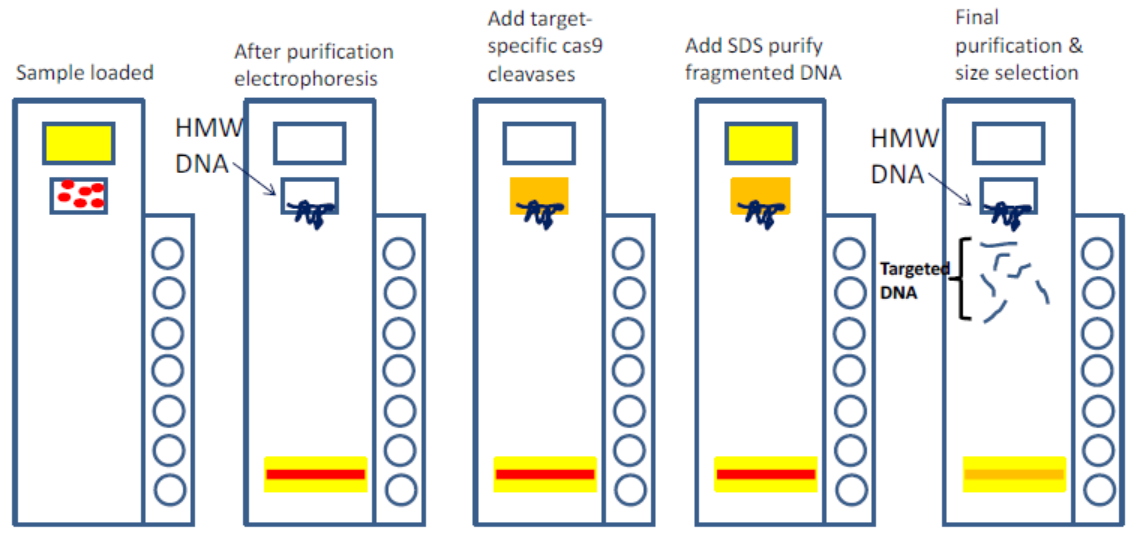
He cast about for a new approach, and found inspiration in the burgeoning CRISPR field. “We came up with this idea that you can cut the flanking region with Cas9 and then use gel electrophoresis to extract only the fragment that you’re looking for,” he says. The method involves RNA-guided Cas9 to make two cuts to pull out the specific region of interest, followed by a size-separation step to remove off-target fragments. It’s geared toward genomic regions that are 50 Kb or larger. Together with Ting Zhu and Chunbo Lou from Tsinghua University, the team began generating custom BACs by combining CATCH with the Gibson assembly to cut the desired piece of DNA and clone it into a vector in a streamlined process.
Since then, Ebenstein and many other labs using CATCH have been broadening the base of applications. It’s particularly attractive for third-gen sequencing platforms; because they typically have lower throughput, “it’s especially beneficial to only probe what you’re interested in and not waste your sequencing depth on regions that are not of interest,” he says. “This is the power of CATCH: no matter how complex the region or what structural variations are in it, if you know the flanking region, you can fish it out and analyze it.”
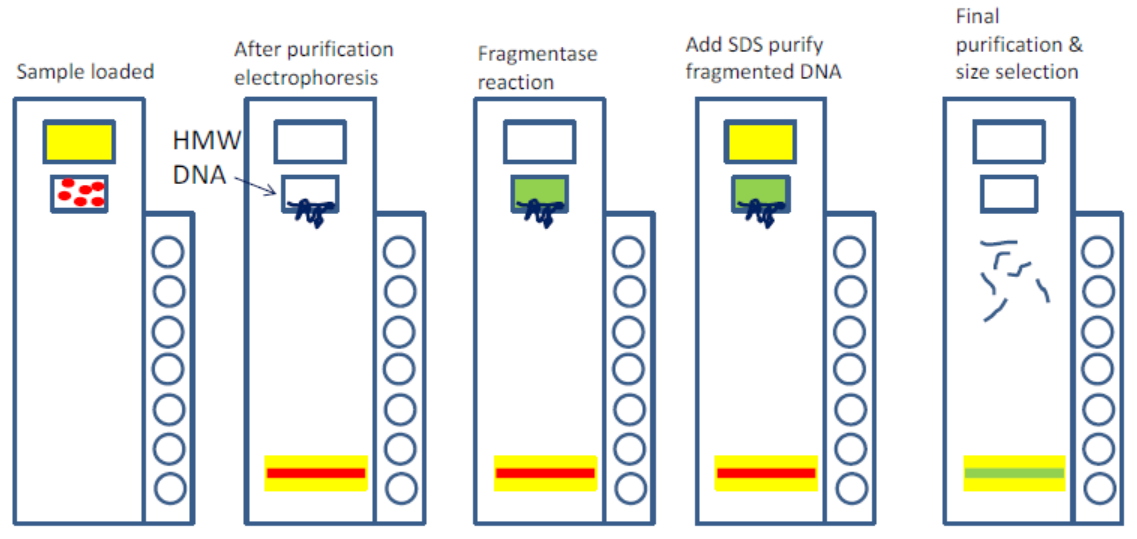
An early drawback with the CATCH protocol was its use of gel electrophoresis, which Ebenstein refers to as “a prehistoric technology.” Size selection is essential for the method, but users must perform the very cumbersome pulsed-field gel electrophoresis technique. That’s where the SageHLS instrument came in. “Sage basically eliminates all of that,” Ebenstein says. The automated platform handles everything inside the gel, and collects size fractions without needing a visible band. “The recovery is phenomenal,” he adds. “You can use a very low amount of starting material and you still get a meaningful amount of DNA for further analysis.”
The protocol for using the SageHLS instrument with CATCH (something we refer to as HLS-CATCH) is still undergoing optimization, with Ebenstein’s team putting the new platform through its paces.
In the meantime, the community continues to push ahead with CATCH. It is already in development in several labs for studies of plants, which have highly repetitive DNA. Ebenstein and others are working to make the protocol robust for use in human genetics as well, targeting important genes such as BRCA1 and BRCA2. He says that the SageHLS instrument will likely be an important factor in those efforts.
How can you tell if CATCH is right for you? Ebenstein has a simple rule: “If you can PCR it, PCR it,” he says. “If you can’t, then you probably need CATCH if you don’t want to go bankrupt.”
This entry was posted on May 18 2017 by Alex, in SageHLS Blog and tagged CATCHPosters and application notes :
Targeted isolation of long genomic DNA molecules
Integrated workflow for Nanopore sequencing of genomic DNA
SageHLS CATCH workflow for targeted Nanopore sequencing
Assembly-based structural variation and haplotypes from targeted sub-Megabase DNA molecules
Assembly of Mb-size genome segments from linked read sequencing of CRISPR DNA targets (bioRxiv)
Targeted sequencing of highly homologous genes using Cas9 digestion
Targeted Mitochondrial DNA Extraction and Enrichment Using the SageHLS System






